How to clone a plasmid really fast and have it work basically every time
Cloning is the most important skill in bioengineering. It does seem like something that is prone to being automated, but it’s not, not yet (in a cheap, and fast way, fast being the most important). The most obvious roadblock to that is how much customization is involved, and DNA synthesis not being fast or cheap enough to compete with e.coli stocks for routine purposes, but I digress.
In the meantime, cloning abilities separate the wheat from the chaff. The faster you can clone the faster you can test ideas. The less effort you can expend on cloning, the more effort you can expend on experiments. Or lunch, or something. The ideal plasmid cloning method is one that requires as little hands-on work as possible, is standard for all routine applications, and produces what you want (clonal, sequenced plasmid) in 1-2 days. Many of the practices that are taught and handed down are just legacy carryover, with no clear purpose.
I use gibson assembly for 95% of the plasmids I clone but have cut out a lot of steps and swapped some for faster, easier steps.
Without further ado:
New technology has changed the optimal way to clone. The tech is nanopore, medium throughput, long read sequencing. What’s important to cloning is that you can sequence whole plasmids for $15. This means you can clone with PCR and deal with the low frequency of mutations introduced without having to sanger sequence your whole plasmid. I almost always clone the entire plasmid with PCR. We have a great CRO near us, called Primordium that does this, major research cities will have their own CRO.
For all cloning purposes, I am a loyalist to Takara Primestar GXL polymerase. It has a low error rate and you can use it at 10 sec/kb. This saves you a lot of time, most polymerases say 1 min/kb. So a 3 kb PCR goes from 180 min extension to 30 min extension. For inserts below ~5 kb I run about 26x cycles of PCR, and above I run about 28x cycles. I put 0.1 ng of plasmid into my PCR. People are sometimes surprised to see how little extra amplification you get later on, more cycles and you’re not increasing yield and just increasing background and smears.

Primers should be designed to have a tm of around 58 on Benchling, or other molecular bio software. If you’re making overlaps make the overlap total around 30 bp. I don’t know the optimal but this always works, won’t hurt to make overlaps longer though. I’ve gotten down to 15 bp overlap to work before, but shorter not so much.
Make the region where the primer binds ~58C, and just extend it to reach 60 bp. In the PCR use a tM of 60C, don’t adjust based on primer tM. PCRing off a plasmid, or fragment is so reliable that if it doesn’t work, I assume I forgot to add a reagent to the reaction or mixed up a template/primer. I can’t remember a time where redesigning primers was necessary. Add the right things and I get a single band of the right size 98% of the time. IDT can send primers as 100 uM liquid now, and not needing to dilute by hand saves some time as well.
PCR:
· 1 uL poly
· 2 uL dNTP
· 5 uL 5X buffer
· 0.1-0.2 uL FW primer (100 uM)
· 0.1-0.2 uL RV primer (100 uM)
· 0.1 ng template
· ddH2O to 25 uL
Normally, I set this up as a master mix w/ only the polymerase, dNTP, buffer, and water. That way you just need to add plasmid + primer to the reaction tube, and can just distribute master mix 24 uL/tube.
If you choose to use a restriction enzyme, you can shorten most digests to 5-15 minutes. I tested a double cutter, and transformed unpurified digests, the background doesn’t change. On this note, I also don’t gel purify digests. The gibson assembly chews back the insert and plasmid and it won’t self-ligate. I promise.

On that note, I also don’t gel purify PCRs. (I’m firmly anti gel extraction, they’re unreliable, and decrease cloning efficiency). You can just go straight from PCR into gibson, 0.5 uL each PCR. Don’t worry about normalizing. If you’re feeling daring you can skip the agarose gel, but I normally don’t. If you use 0.1-1 ng of plasmid as template in the PCR, it will be so dilute in the final gibson mix that it won’t contribute much background. No need to dpnI either.
To check PCRs, I like to use an agarose gel with a lithium acetate boric acid buffer instead of TAE. This lets you run at 300V versus ~100V for a TAE gel. With these gels it’s better to load less DNA, the gels are fast but the bands are prone to getting blobby if you load too much. 3 uL is enough, and the bands will stay clean. I normally run for 15 minutes and pour the gel while the PCR is going. Here is a side-by-side.

And a gel where I loaded only 3 uL of a PCR.
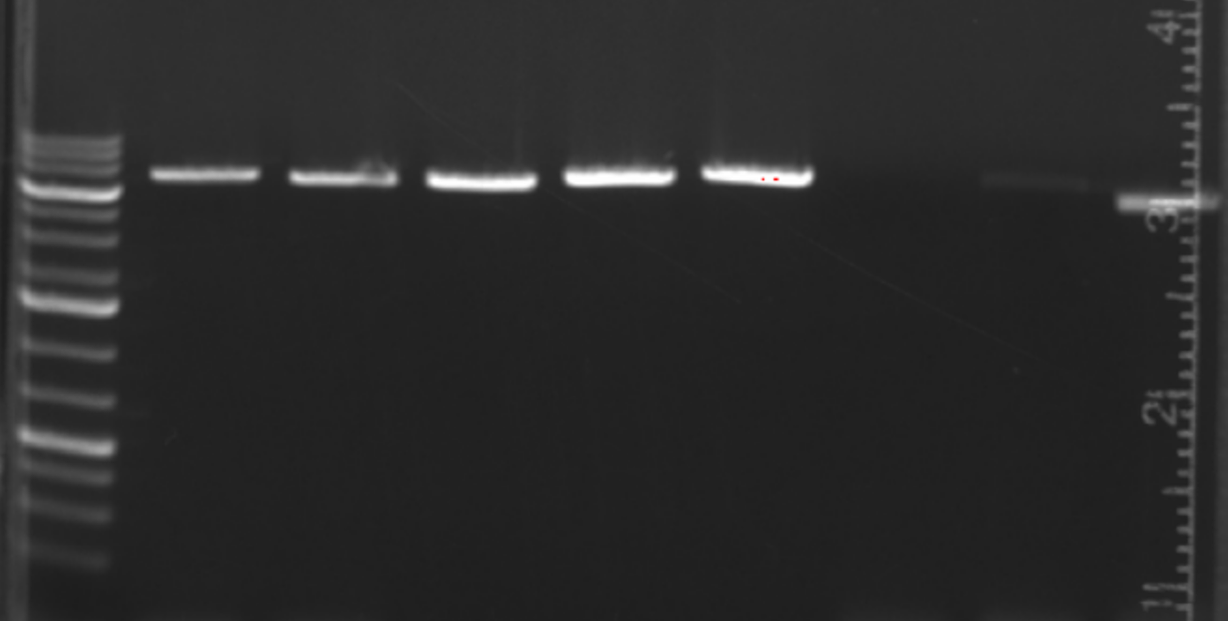
From a functional PCR, set up the Gibson as such:
· 0.5 uL insert fragment PCR
· 0.5 uL insert fragment PCR
· 0.5 uL vector fragment PCR
· ddH2o to 2.5 uL
· 7.5 uL Gibson MM (prep in house, commercial mixes are garbage)
15 at 50C minutes is fine for most gibson assemblies. You can extend to 1 hr and it does increase the efficiency, but generally you don’t need it. I still get dozens of colonies on the gibson, and 0-4 on the background vector only control. Only one has to be good. I also will include a control which is just the vector amplification. Bacteria can do some weird stuff to get that antibiotic resistance.
With chem comp transforming, I normally go by the book. Ampicillin you can plate right after heat shock but normally if the actual cloning is done quickly, it’s done by 2, and I’m not rushing out the door.
One trick I use often is PCR assembly. It’s not necessary for 2- and 3-part gibsons, but once you get to 5 and above its useful because efficiency starts to drop dramatically around here. Basically, you can just stick multiple gibson fragments together by only including the outermost primers. Because the fragments already have overlaps from PCR 1, they will stick together and only pieces which have both ends (primer binding sites) will be exponentially amplified.
If I need to PCR assemble, I do gel extract the individual fragments to purify out primers. I’ve assembled up to 3 pieces in one reaction, but these reactions can be messy and gel purifying might be necessary, often it isn’t though.
· 1 uL GXL poly
· 2 uL dNTP
· 5 uL 5X buffer
· 0.1-0.2 uL FW primer (100 uM)
· 0.1-0.2 uL RV primer (100 uM)
· 5 ng fragment 1
· 5 ng fragment 2
· ddH2O to 25 uL
Here is what a typical gibson looks like (top two are gibson), bottom two are control. I’ve gotten up to 4 parts unstitched to work with this unpurified PCR method.

Day 1 is the bulk of the work. With my tricks, we’ve gone from:
· 30 cycle Q5 PCR – 3 hrs
· Plasmid digest – 1 hour (concurrent w/ PCR)
· TAE agarose gel – 45 min
· Gel extraction – 45 min
· Gibson – 1 hour
· Transformation – 1.5 hrs
· Total – 7 hours, hands on – 2.5-3 hours
To:
· GXL PCR – 1 hr (all pieces)
· Lithium gel – 15 min
· Gibson – 15 min
· Transformation – 1.5 hrs
· Total – 3 hours, hands on - 45 mins
Our primers are either delivered at 10 AM, or 1 PM. This basically guarantees that you can make the construct the same day you receive, which is one day after ordering. It also comfortably gives you enough time to repeat a PCR if you think you made a mistake setting it up. With a slower polymerase might get pushed to the next day.
The next day you’ll have colonies on your gibson, and very few on your control. There are two options from this point to sequence your plasmid same day. Most of the time I do colony PCR, it scales far easier than minipreps. I have a set of primers which amplifies nearly the entire plasmid. GXL poly works well for this, I’ve amplified plasmids up to 10 kb with no problem.
· Pick colonies into 10 uL ddH2O.
· Mix and use 1 uL as template
· Save the rest, to inoculate a full miniprep later
· Set up reaction as before, 28x cycles
Normally, you can just set up a master mix for all the plasmids w/ the same primers bc certain plasmid regions are common (origin of replication, antibiotic resistance, etc). Run the PCR, add 3 uL of SDS free 10X Loading dye (I use a ficoll based one), run the 15 min LiAc gel. Check size of bands. Inoculate ones with the right size into LB/antibtiotic.

This PCR + LD mix can be sent to primordium directly with no purification for the same price as a purified plasmid. Any errors in the colony PCR will average out, but any errors in the actual clonal plasmid will appear. The sequencing does fail every so often, but in my hands its only ~1/20 reactions. If I’m transforming yeast w/ the construct I’ll often just use the PCR as the transforming DNA, and just save the bacteria as a glycerol stock.
The alternative to colony PCR is picking colonies as early as possible and miniprepping at the end of the day to make the sample submission cutoff, but personally I don’t like leaving stuff to the very end of the day.
The method of cloning I was taught required picking colonies on day 1- miniprepping, restriction enzyme digesting, and submitting for sanger on day 2 – getting results on day 3. My method cuts a day out of that cycle, and saves a lot of hands on work doing dozens of wasted minipreps and setting up dozens of sanger reactions.
This is a recent example of how cloning normally goes.
I receive primers at 11:00 on a wednesday, and rush to set up my PCRs before a 12-1:30 meeting. I’m making 9x constructs with 3 different backbones, 6x 2 piece gibsons, 2x 3 piece, and 1x 4 piece gibsons. Post meeting I run a 15 minute fast gel.

All look good, so I set up my gibsons as usual, 0.5 uL of each fragment, water up to 2.5 uL, 7.5 uL gib mix. Transform into Top10 and a special ccbd survival strain for a couple.
Next day, all gibsons have more than the vector only control, the 4x part gibson has about 5x more colonies than some 2x gibsons, weirdly. I set up my screening reactions with colony PCR, again rushing before a 12:00 meeting, and balancing with preparing transformation competent yeast for the next day and reading out another experiment by flow.

All look good (save the ugly gel), and the vector only control is largely not the parent vector, but a weird re-ligation. I submit 2-3x unpurified PCRs per construct for sequencing depending on the ratio vs. the vector only control. I also inoculate all 4x that were PCR’d into a 48 well block for outgrowth in LB+Amp.

The next morning all sequencing reactions have worked, and I align to the desired construct. 100% are the correct construct, and each construct has at least 1x clone with no SNPs in the gibson junctions, or SNPs introduced by PCR. I glycerol stock and miniprep those and use them in a transformation on Friday.

That’s basically it. I’ve been cloning like this for a while now and it’s at least as, if not more, reliable than the more “rigorous” method (above is a side-by-side with top row unpurified, and bottom purified) and produces exactly what you want with not much thinking, hands-on work, and in substantially less time.